


Методика контроля содержания свободной глутаминовой кислоты в пищевой продукции
- 30.06.2023 12:48
В настоящее время в пищевой промышленности широко применяются усилители вкуса и аромата. Данные пищевые добавки призваны влиять на вкусовые рецепторы человека. Многие из них не только усиливают вкус продукта, но и влияют на его запах. Усилители вкуса и аромата применяют при производстве мясо- и рыбопродуктов, продуктов переработки овощей, грибов, соусов, кетчупов, продуктов быстрого приготовления, бульонных кубиков, вкусо-ароматических смесей для обсыпки чипсов, орехов, кондитерских изделий.
Одним из самых распространённых усилителей вкуса и аромата, применяемых в пищевой промышленности, является глутаминовая кислота (ГК) (Е620) и ее соли (Е621-Е625). Однако чрезмерное потребление продукции с данными добавками может негативно сказаться на здоровье человека, вызвать головную боль, повысить артериальное давление и уровень инсулина [1].
Содержание ГК и ее солей в пищевой продукции регулируется требованиями технического регламента Таможенного союза ТР ТС 029/2012 «Требования безопасности пищевых добавок, ароматизаторов и технологических вспомогательных средств» [2], который устанавливает предельное содержание добавок Е 620–Е 625 в пищевых продуктах на уровне 10 г/кг (в пересчете на глутаминовую кислоту).
Все выше изложенное обуславливает необходимость проведения строгого контроля содержания свободной ГК в пищевой продукции, произведенной не только на территории Республики Беларусь, а так же государств-членов ЕАЭС.
Высокоэффективная жидкостная хроматография является одним из основных методов исследования, применяемым при анализе ГК в пищевой продукции [3–8], и лежит в основе ряда аттестованных методик. Однако методики, действующие на территории государств-членов Евразийского экономического союза, обычно включают в себя исследование содержания общей ГК (связанной в белке и свободной). Кроме того, имеющиеся методики по определению свободной ГК не охватывают весь спектр пищевой продукции [4, 6-7]. В связи с этим целью данной работы явилась разработка методики, позволяющей быстро, с высокой надежностью и достоверностью определять свободную ГК во всех видах пищевой продукции.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы, материалы, оборудование
В качестве стандартного образца использовали L-(+)-глутаминовую кислоту (≥ 99,0%, Sigma-Aldrich).
Использовали ацетонитрил для ВЭЖХ (Fisher Chemical), метанол для ВЭЖХ (Acros Organics), кислоту солянуюя (х.ч), натрий фосфорнокислый двузамещенный 12-водный (х.ч), тетраборат натрия (х.ч), азид натрия (х.ч), кислоту борную (х.ч), натрия гидроокись (х.ч), 3-меркаптопропионовую кислоту (≥ 99,0%, Sigma-Aldrich), ортофталевый альдегид (Glentham Life Sciences). Условия хроматографирования оптимизировали на колонке Eclips Plus C18 длиной 100 мм, внутренним диаметром 4,6 мм и с зернением сорбента 3,5 мкм (Agilent Technologies, Германия). Использовали ультразвуковую баню Sonorex super RK 103H (Bandbelin, Германия), центрифугу охлаждаемую 3-18К (Sigma, Германия), иономер (рН-метр) с диапазоном измерения от 0,00 до 14,00 (Thermo, Польша). Количественное определение ГК проводили с помощью жидкостного хроматографа, оснащенного диодно-матричным и флуоресцентным детекторами Agilent 1260 (Agilent Technologies, США-Германия).
Приготовление градуировочных растворов
Градуировочные растворы ГК массовыми концентрациями 15,0; 30,0; 45,0; 60,0 мкг/см3 готовили из основного стандартного раствора ГК концентрацией 1,0 мг/см3, полученный в результате растворения навески ГК в растворе соляной кислоты концентрацией 0,1 моль/дм3 с учетом содержания основного вещества в стандартном образце.
Градуировочный раствор ГК массовой концентрацией 5,0 мкг/см3 готовили из стандартного раствора ГК концентрацией 100 мкг/см3, полученный путем разбавления основного стандартного раствора ГК концентрацией 1,0 мг/см3.
Приготовление боратного буферного раствора
В стакан вместимостью 250 см3 помещали 100 см3 2 % раствора борной кислоты и приблизительно 60 см3 2 % раствора гидроксида натрия, устанавливали рН буферного раствора 10,2 при помощи 2 % раствора гидроксида натрия.
Приготовление раствора дериватизирующего реагента
В центрифужную пробирку вместимостью 50 см3 помещали (0,200 +- 0,003) г орто-фталевого альдегида, добавляли 20 см3 боратного буферного раствора и 0,2 см3 3-меркаптопропионовой кислоты, затем помещали пробирку на электровстряхиватель на 60 мин.
Приготовление дериватизированных градуировочных растворов ГК
В мерную колбу вместимостью 10 см3 помещали 1 см3 боратного буферного раствора с рН=10,2, 0,5 см3 градуировочного раствора ГК, 0,3 см3 раствора дериватизирующего реагента, содержимое перемешивали. Затем в колбу приливали 1 см3 раствора HCl молярной концентрации 0,1 моль/дм3, доводили объем дистиллированной объем до метки на колбе и перемешивали.
Условия хроматографирования
Идентификацию ГК проводили по времени удерживания при длине волны поглощения 340 нм и длине волны эмиссии 450 нм.
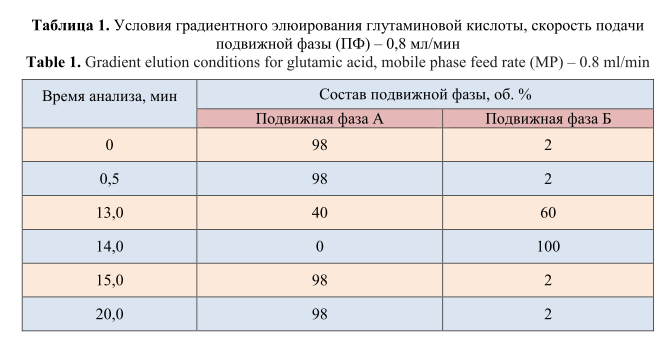
Условия градиентного элюирования ГК представлены в таблице 1.
Подвижная фаза А: фосфатно-боратный буферный раствор с рН=8,2: 0,14% раствор натрия фосфорнокислого двузамещенного и 0,38% раствор натрия тетраборнокислого.
Подвижная фаза Б: метанол:ацетонитрил:вода (45:45:10 об.%).
Подготовка анализируемых образцов
Экстракция ГК из пищевой продукции (хлебобулочные, плодоовощные, мясные, рыбные продукты, специи) с массовой долей жира менее 40%
От 0,5 до 2,0 г пробы помещали в мерную колбу вместимостью 100 см3, добавляли 50 см3 раствора НСl молярной концентрацией 0,02 моль/дм3, интенсивно встряхивали и помещали в ультразвуковую баню при температуре от 20 до 25оС на 10 мин (экстракции ГК из пищевой продукции с массовой долей жира более 20% проводится в ультразвуковой бане при температуре от 50 до 60оС в течение 10 мин, при этом колбу с пробой периодически интенсивно встряхивают (от 3 до 4 раз в течение 10 мин). Затем содержимое колбы доводили раствором НСl молярной концентрацией 0,02 моль/дм3 до метки на колбе и перемешивали. 10 см3 экстракта переносили в центрифужную пробирку вместимостью 15 см3 и центрифугировали при частоте вращения 10 000 об/мин при температуре от 5°С до 10°С в течение 10 мин. При необходимости, верхний липидный слой центрифугата отбрасывали. Далее проводили дериватизацию ГК, содержащейся в экстракте, согласно процедуре, описанной при приготовлении градуировочных растворов. В данном случае вместо 0,5 см3 градуировочного раствора ГК используется 0,5 см3 экстракта.
Экстракция ГК из пищевой продукции с массовой долей жира более 40 %
Отбирали среднюю пробу массой от 0,5 до 2,0 г, помещали в центрифужную пробирку вместимостью 50 см3, добавляли 30 см3 раствора НСl молярной концентрацией 0,02 моль/дм3 и 10 см3 гексана, встряхивали в течение 5 мин. Далее содержимое пробирки отстаивалось в течение 10 мин для достижения межфазного равновесия. При необходимости (отсутствие четкой межфазной границы, образование эмульсии) пробу центрифугировали при частоте вращения 10000 об/мин в течение 5 мин. Гексановую фракцию отбрасывали, водный слой количественно с помощью раствора НСl молярной концентрацией 0,02 моль/дм3 переносят в мерную колбу вместимостью 100 см3 доводят объем раствором НСl молярной концентрацией 0,02 моль/дм3 до метки и перемешивали. 10 см3 экстракта переносили в центрифужную пробирку вместимостью 15 см3 и центрифугировали при частоте вращения 10 000 об/мин при температуре от 5°С до 10°С в течение 10 мин. При необходимости верхний липидный слой центрифугата отбрасывали. Далее проводили дериватизацию ГК, содержащейся в экстракте, согласно процедуре, описанной при приготовлении градуировочных растворов. В данном случае вместо 0,5 см3 градуировочного раствора ГК используется 0,5 см3 экстракта.
Расчет содержания ГК и ее солей в анализируемых образцах
Массовую долю L-(+)-глутаминовой кислоты Х, мг/кг, вычисляют по формуле:
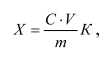
где С – массовая концентрация L-(+)-глутаминовой кислоты, найденная по градуировочному графику, мкг/см3;
m – масса навески, г;
V – объем разведения, см3;
К – коэффициент разбавления.
Если площадь хроматографического пика L-(+)-глутаминовой кислоты в анализируемой пробе находится в диапазоне градуировочного графика, то К=1.
Если площадь хроматографического пика L-(+)-глутаминовой кислоты в анализируемой пробе превышает верхнюю точку градуировочного графика, то необходимо провести разбавление анализируемой пробы. Коэффициент разбавления в данном случае вычисляют по формуле:
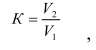
где V1 – аликвота раствора пробы, взятая для разбавления, см3;
V2 – объем пробы после разбавления, см3.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Оптимизация условий ВЭЖХ определения ГК
Обращенно-фазовый вариант ВЭЖХ с фотометрическим или флуоресцентным детектированием является наиболее экспрессным и доступным при анализе аминокислот.
Так как в большинстве молекул аминокислот, в том числе и ГК отсутствуют хромофорные группы (рисунок 2), они, как правило, слабо поглощают ультрафиолетовый (УФ) и видимый свет, не обладают собственной флуоресценцией, соответственно чувствительность их определения с помощью спектрофотометрических и флуориметрических детекторов невысока. Для достижения удовлетворительного разделения и детектирования аминокислоты, как правило, переводят в гидрофобные соединения, поглощающие УФ излучение, либо сильно флуоресцирующие производные, т.е. проводят предколоночную или постколоночную дериватизацию [3-14].
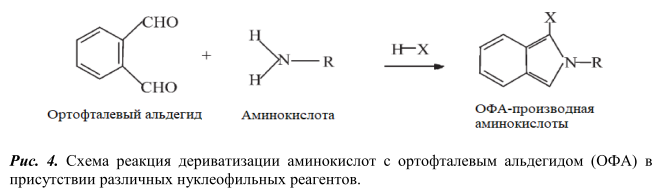
В качестве реагентов для дериватизации применяют диметиламиноазобензосульфонил хлорид (ДАБС) [3], фенилизотиоционат [6, 9, 10], ортофталевый альдегид с различными нуклеофильными агентами (цианидом калия, сульфитом натрия, 3-меркаптпропионовая кислота, N-ацетил-L-цистеином, 9-фторенилметил хлорформиатом) [7, 11-12], 4,7-фенантролин-5,6-диона (фанхинона) [13], нингидрин [14] и др.

При этом условия проведения реакции, полнота ее протекания, а также устойчивость продуктов дериватизации могут сильно различаться в зависимости от используемого дериватизирующего агента. Соответственно условия разделения полученных производных аминокислот на хроматографической колонке также будут в значительной степени определяться физико-химическими свойствами соединений, участвующих в дериватизации.
Так как основными интерферирующими компонентами пробы пищевой продукции при определении ГК являются аминокислоты, то в данной работе было изучено разделение смеси 17 аминокислот на обращеннофазных хроматографических колонках при использовании разных дериватизирующих реагентов: диметиламиноазобензосульфонил хлорида (ДАБС) и орто-фталевого альдегида (ОФА). Схемы протекания реакций дериватизации представлены на рисунках 3 и 4 соответственно. Для детекции аминокислот использовали диодноматричный детектор.
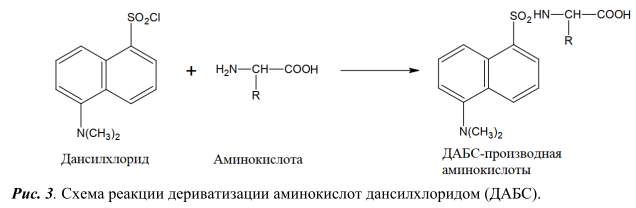
Условия проведения реакции дериватизации стандартной смеси аминокислот c помощью ДАБС-реагента и условия ВЭЖХ анализа полученных производных аналогичны условиям, описанным в методике [3]. Реакция дериватизации протекает при 70°С в течение 12 мин. Разделение проводили на хроматографической колонке HyperClone ODS (C18) (250 мм x 4 мм х 5 мкм) в градиентном режиме подачи подвижной фазы, длина волны поглощения 436 нм. Хроматограмма стандартной смеси 17 аминокислот концентрацией 5 нмоль/см3 на рисунке 5.
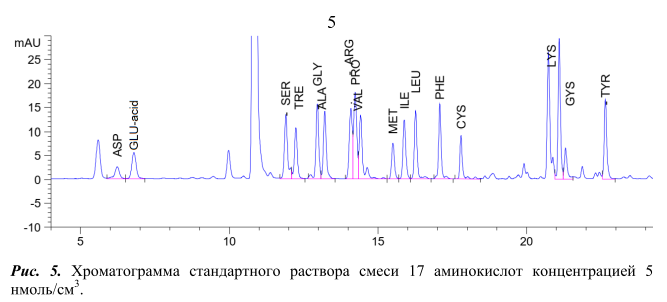
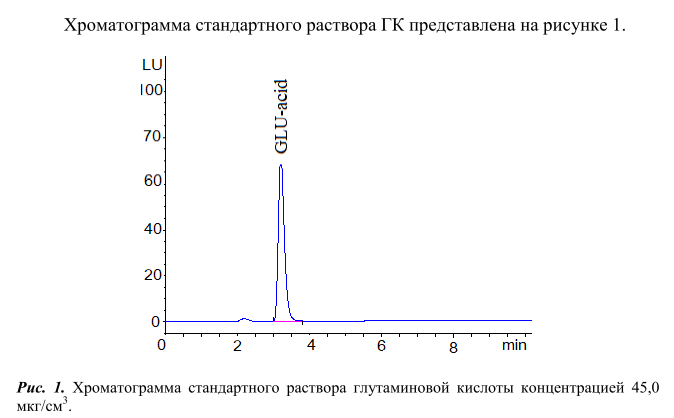
Из хроматограмы видно, что ДАБС-производное глутаминовой кислоты (GLU-acid) выходит из хроматографической колонки на 7 минуте. Пик данного соединения хорошо отделен от пиков ДАБС-производных других аминокислот.
Дериватизацию аминокислот орто-фталевым альдегидом в присутствии нуклеофильного агента 3-меркаптопропионовой кислоты проводили согласно методике, описанной в работе [12]. Реакция дериватизации протекает при 25°С, 5 мин. Разделение проводили на хроматографической колонке Agilent Poroshel C18 (100 мм х 4,6 мм х 2,7 мкм) в градиентном режиме подачи подвижной фазы, длина волны поглощения 340 нм.
Как и в первом случае, OФA-производное глутаминовой кислоты выходит из колонки одним из первых (на 4-ой минуте), пик данного соединения также хорошо отделяется от интерферирующих пиков производных других аминокислот. Поэтому описанные в работах [3] и [12] условия являются подходящими для решения задачи по определению свободной (добавленной) ГК в пищевой продукции.
Следует отметить, что использование ОФА в качестве дериватизирующего агента является предпочтительным по сравнению с ДАБС, так как условия протекания реакции более мягкие (T=25°С, 5 мин), что позволяет автоматизировать данный процесс с помощью современных ВЭЖХ систем: проводить реакцию дериватизации в виале непосредственно перед вводом пробы в хроматограф. Анализ становится менее трудоемким и времязатратным.
Получаемые ОФА-производные аминокислот являются флуоресцирующими соединениями, что дает возможность использовать флуоресцентный детектор для определения ГК и тем самым увеличить чувствительность методики ее определения, а также уменьшить влияние примесей в пробе путем увеличения кратности ее разбавления. Так как ОФА-производная глутаминовой кислоты выходит из хроматографической колонки одной из первых, то использование градиентного режима элюирования (постепенное увеличения доли органического компонента подвижной фазы) позволяет быстро очистить колонку от интерферирующих примесей, при этом также наблюдается гашение флуоресценции ОФА-производных других аминокислот, что также способствует повышению селективности методики.
Оптимизация условий пробоподготовки
В зависимости от метода анализа и природы анализируемого пищевого продукта в литературных источниках описаны различные способы подготовки пробы для определения содержание ГК. Пищевые продукты могут содержать значительное количество ГК природного происхождения, как в свободных, так и в связанных формах. Свободные формы ГК выделяют из образцов с помощью экстракции.
В работах [7, 9, 11, 15] для экстракции ГК используются спиртовые, водно-спиртовые растворы и растворы соляной кислоты различной концентрации. Представляло интерес изучить влияние состава экстрагента на полноту извлечения ГК из пищевой матрицы. Для этого в пробы завтрака сухого на основе картофеля, не содержащие внесенной ГК, была внесена данная добавка в количестве 5 г/кг.
В качестве экстрагентов использовали дистиллированную воду, растворы НCl с концентрацией 6 моль/дм3 и 0,02 моль/дм3 , водно-этанольный раствор (соотношение растворителей 1:1). Для дериватизации и количественного
определения внесенной ГК использовали ортофталевый альдегид и нуклеофильный агент 3-меркаптопропионовую кислоту.
В результате проведенных исследований было установлено, что состав исследуемых экстрагирующих смесей не оказывает существенного влияния на выделение добавленной ГК из данной пищевой матрицы. Степень извлечения
составила 92 – 110% от внесенного количества. При этом наибольшие значения степени извлечения (98 – 110%) установлены при использовании в качестве экстрагентов растворов соляной кислоты. Использование 6 М раствора соляной кислоты приводит к завышению результатов анализа (степень извлечения > 100%). Это объясняется частичным гидролизом белка, в результате чего определяется не только внесенная ГК, но и ГК, высвободившаяся из белка при гидролизе. Поэтому оптимальным экстрагирующим раствором является 0,02 М соляная кислота. Степень извлечения при ее использовании составила 98%.
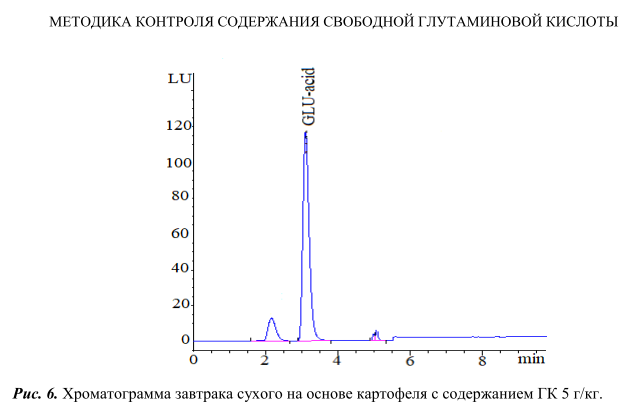
На рисунке 6 представлена хроматограмма, полученная при анализе завтрака сухого на основе картофеля с использованием данного экстрагента. Как видно из рисунка 6, пик ГК четкий, хорошо отделен от других пиков, практически отсутствуют пики примесей на хроматограмме, что позволяет проводить количественное определение данной пищевой добавки на регламентируемом уровне.
Валидация методики определения свободной ГК в пищевой продукции
Валидация методики определения свободной глутаминовой кислоты в пищевой продукции проводилась согласно [16–21]. Для установления линейности диапазона измерений строили градуировочный график, откладывая по оси абсцисс концентрацию глутаминовой кислоты в градуировочном растворе (мкг/см3 ), а по оси ординат площадь пика (LU⋅s). Расчет градуировочного графика проводился методом наименьших квадратов. Критерием линейности являлся коэффициент корреляции R 2 , который был не менее 0,99 (рисунок 7).
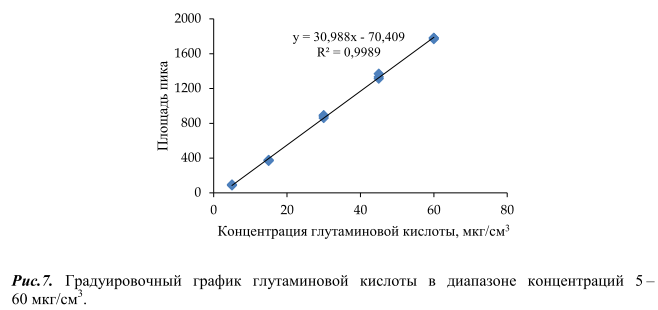
Установленный диапазон определяемых концентраций глутаминовой кислоты составил 0,25 – 100 г/кг продукта. Нижний предел определения найден, исходя из наименьшего значения концентрации градуировочных растворов,
величины навески продукта и конечного объема экстракта.
Статистические данные для оценки прецизионности получены по результатам анализа образцов рабочих проб следующих продуктов:
– сухарики ржаные (1 уровень – 500 мг/кг),
– чипсы (2 уровень – 3000 мг/кг),
– специи (6 уровень – 95000 мг/кг),
а также проб с добавками ГК:
– консервы рыбные для детского питания (3 уровень – 5000 мг/кг),
– мясо (свинина) (4 уровень – 6500 мг/кг),
– томатная паста (5 уровень – 75000 мг/кг).
Проведено по 9 определений (n=2) для каждого образца, выполненных с двумя изменяющимися факторами: время, оператор. Статистическая обработка полученных результатов, показала, что они достоверны при доверительной
вероятности 95%. Значения величины относительного стандартного отклонения повторяемости и относительного доверительного интервала среднего значения не превышала критериев приемлемости – 5%, что свидетельствовало о
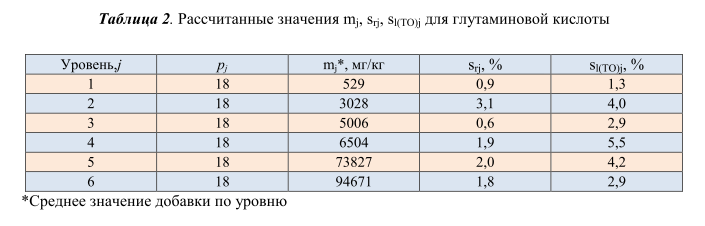
прецизионности методики в условиях повторяемости. Рассчитанные на основании полученных данных относительные стандартные отклонения овторяемости (s r ), промежуточной прецизионности (s l(TO) ) для различных групп продуктов представлены в таблице 2.
Установленные значения s r и s l(TO) определены по наибольшим рассчитанным значениям: s r =3,1%, s l(TO) =5,5%. Пределы повторяемости r и промежуточной прецизионности R I(TO) равны соответственно 8,7% и 15,3%.
Величину степени извлечения (точность) метода получали как отношение результата измеренного содержания ГК в пробе с добавкой к расчетному количеству ГК в пробе с добавкой в соответствии с экспериментальными
данными.
Анализ проб с добавкой ГК показал, что степень извлечения ГК для всех матриц составила не менее 95%.
Относительная стандартная неопределенность измерения свободной ГК в пищевой продукции, согласно разработанной методике, включала: неопределенность, обусловленную случайными факторами (фактор повторяемости), неопределенность, обусловленную смещением метода (извлечение); неопределенность, обусловленную построением и использованием градуировочной характеристики и неопределенность, обусловленную подготовкой пробы, и составила 26,3% (таблица 3).

Практическое использование разработанной методики определения свободной ГК в пищевой продукции
Разработанная и валидированная методика применена для контроля содержания свободной ГК в пищевой продукции. Данной методикой проанализированы 146 образцов пищевой продукции, отобранной в торговой сети Республики Беларусь. Вся продукция условно разделена на 5 групп, в зависимости от типа пищевой матрицы. Практически все проанализированные образцы содержали свободную ГК в количестве от 0,3 г/кг до 5,6 г/кг. Результаты исследований представлены в таблице 4.
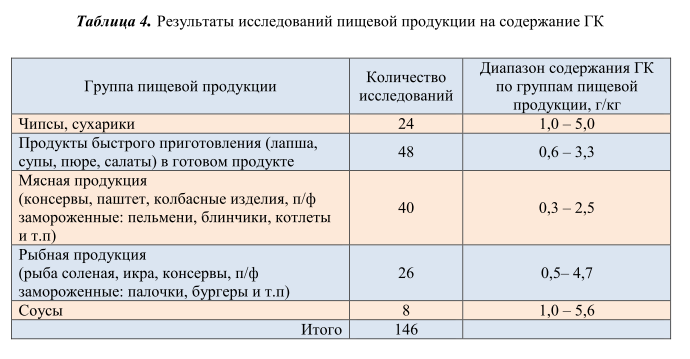
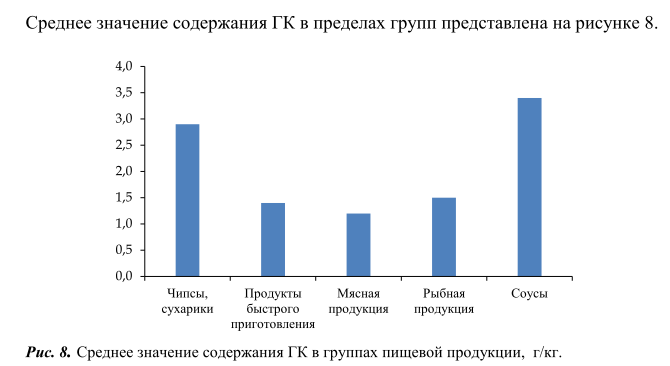
Из рисунка 7 видно, что максимальное количество свободной ГК содержалось в таких продуктах, как чипсы, сухарики и соусы, минимальное количество свободной ГК обнаружено – в мясной и рыбной продукции. Следует отметить, что содержание свободной ГК во всех исследованных образцах ниже максимально допустимого уровня, установленного в требованиях технического регламента Евразийского экономического союза [1].
ВЫВОДЫ
В результате проведенных исследований разработаны оптимальные условия пробоподготовки и хроматографического анализа для количественного определения свободной ГК в пищевой продукции, что позволило разработать и валидировать простую в исполнении методику определения ГК с помощью ВЭЖХ–ФЛД. Диапазон количественного измерения методики составляет 0,25–100 г/кг. Максимальная расширенная неопределенность полученных результатов не превысила 26,3% на нижнем уровне диапазона измерения. Данная методика позволяет с высокой точностью и чувствительностью определять ГК и осуществлять контроль за ее содержанием во всех видах
пищевой продукции.
Работа выполнена в рамках ОНТП «Гигиеническая безопасность», задание 02.08 «Разработать и внедрить метод оценки риска здоровью, ассоциированного с содержанием усилителей вкуса и аромата в пищевых продуктах (на примере глутаминовой кислоты и ее солей)».
Список литературы:
1. Mortensen A., et al. (2017). Re-evaluation of glutamic acid (E 620), sodium glutamate (E 621), potassium glutamate (E 622), calcium glutamate (E 623), ammonium glutamate (E 624), and magnesium glutamate (E 625) as food additives. EFSA Journal, 15(7), 90 pp.
2. ТР ТС 029/2012. Требования безопасности пищевых добавок, ароматизаторов и технологических вспомогательных средств. Минск: Госстандарт, 2014. 272 с.
3. МВИ.МН 1363-2000. Метод определения аминокислот в продуктах питания с помощью высокоэффективной жидкостной хроматографии. Минск: Республиканский научно-практический центр по экспертной оценке качества и безопасности продуктов питания, 2000. 23 с.
4. ГОСТ 32195-2013 (ISO 13903:2005). Корма, комбикорма. Метод определения содержания аминокислот. Минск: Госстандарт, 2016. 24 с.
5. ГОСТ 34230-2017. Продукция соковая. Определение свободных аминокислот методом высокоэффективной жидкостной хроматографии. Минск: Госстандарт, 2019. 20 с.
6. ГОСТ 34448-2018. Мясо и мясные продукты. Метод определения L-(+)-глутаминовой кислоты. Минск: Госстандарт, 2019. 20 с.
7. Руденко А.О., Карцова Л.А., Снарский С.И. (2010). Определение важнейших аминокислот в сложных объектах биологического происхождения методом обращенно-фазовой ВЭЖХ с получением фенилтиогидантоинов аминокислот. Сорбционные и хроматографические процессы, 10(2), 223–230.
8. Tsvetkova D., Pencheva I., Obreshkova D. (2010). Identification and determination of L-glutamic acid and L-arginine by HPLC in food additives. Acta Pharmaceutica Sciencia, 52(2),219–228.
9. Степанов К.В., Пирогов А.В., Дикунец М.А., Шпигун О.А. (2005). Получение фенилтиогидантоинов аминокислот для количественного анализа аминокислотного состава белков методом капиллярного электрофореза. Вестник Московского университета. Серия 2. Химия, 46(6), 395–399.
10. Чернобровкин М.Г., Кольцова Н.В., Шепелев Б.Н. (2004). Определение аминокислот в препарате «Элтацин». Фармация, 53(5), 18–20.
11. Бекетов В.И., Воронина Р.Д., Зоров Н.Б. (2012). Флуориметрическое определение аминокислот и фотохимическая устойчивость продуктов их реакции с ортофталевым альдегидом под воздействием мощного импульсного лазерного излучения. Вестник Московского университета. Серия 2. Химия, 53(4), 228–233.
12. Jajic I., Krstovic S., Glamocic D., Jaksic S., Abramovic B. (2013). Validation of an HPLC method for the determination of amino acids in feed. Journal of the Serbian Chemical Society, 78 (6), 839–850.
13. Gatte R., Gioia M.G., Pieta A.M. (2002). Phanquenone: a useful fluorescent pre-chromatographic derivatization reagent for liquid chromatographic analyses of aminoacid dosage form. Analytica Chimica Acta, 1(2), 11–20.
14. Krishna V.N., et al. (2010). Analysis of monosodium L-Glutamate in food products by High-Performance thin layer chromatography. Journal Young Pharm, 2(3), 297–300.
15. Afraa A, Mounir A, Zaid A (2013). Colorimetric determination of monosodium glutamate in food samples using L-glutamate oxidase. Clinical Journal of Applied and Environmental Biology, 19(6), 1069-1072.
16. СТБ ИСО 5725–2–2002 Точность (правильность и прецизионность) методов и результатов измерений в 6 ч. – Ч. 2: Основной метод определения повторяемости и воспроизводимости стандартного метода определений. Минск: Госстандарт, 2002. 42 с.
17. СТБ ИСО 5725–3–2002 Точность (правильность и прецизионность) методов и результатов измерений в 6 ч. – Ч. 3: Промежуточные показатели прецизионности стандартного метода измерений. Минск: Госстандарт, 2002. 36 с.
18. СТБ ИСО 5725–4–2002 Точность (правильность и прецизионность) методов и результатов измерений в 6 ч. – Ч. 4: Основные методы определения правильности стандартного метода определений. Минск: Госстандарт, 2002. 32 с.
19. СТБ ИСО 5725-6-2002 Точность (правильность и прецизионность) методов и результатов измерений. Часть 6. Использование значений точности на практике. Минск: Госстандарт, 2002. 48 с.
20. ГОСТ 8.010-2013 Государственная система обеспечения единства измерений (ГСИ). Методики выполнения измерений. Основные положения. Москва: Стандартинформ, 2019. 99 с.
21. СТБ ISO 21748-2019 Руководство по использованию оценок повторяемости, воспроизводимости и правильности при оценивании неопределенности измерений. Минск: Госстандарт, 2019. 40 с.






























| понедельник | вторник | среда | четверг | пятница | суббота | воскресенье |
|---|---|---|---|---|---|---|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
